I was very pleased to listen to your State of the Union address and learn of your interest in Precision Medicine. As I am sure you know, this has led to a number of commentaries about what this term actually means (here, here, here). I would like to provide yet another perspective, this time from someone who has practiced clinical medicine, led academic research teams and currently works in the pharmaceutical industry.
Let me start by acknowledging that I know very little about your plan, but that is because no plan has been announced. However, that inconvenient fact should not prevent me from forming a very strong opinion about what you should do. Similar behavior is observed in politics (which you know well) and sports radio (see for example “Deflate-gate”). So here it goes…
I want to clarify my definition of “precision medicine” (see here for my previous blog on how this is different from “personalized medicine”). In the simplest of terms, precision medicine refers to the ability to classify individuals into subpopulations based on a deep understanding of disease biology. Note that this is different than what clinicians normally practice, which is to classify patients based on signs and symptoms (which can be measured by clinicians as part of routine clinical appointments).…
Recently I was asked by the American Society of Human Genetics (ASHG) to provide my perspective on career and professional development in genetics (see here). At about the same time, I read the book “How Google Works”, by Google Executive Chairman and ex-CEO Eric Schmidt and former SVP of Products Jonathan Rosenberg. A very creative slide deck accompanies the book, which is definitely worth a few minutes of your time (here).
Both got me thinking about opportunities in the pharmaceutical industry for genetic graduate students. Here are a few thoughts based on the outline from the Google slide deck.
What is different now?
In human genetics, large-scale genotyping and sequencing is unlocking the inherited basis of most complex and rare traits in the ideal model organism, humans. This is very different than it was just a few years ago. But there is more: this is happening at a scale that will not likely stop until most humans on the planet have their genome sequenced. Like the “Internet of things”, there will soon be a “Genomes of things”, in which our genomes will be connected to all sorts of data – electronic health records, wearable technology, portable blood monitoring, etc.…
Welcome to our first blog of 2015 on genetics/genomics for drug discovery. After a nice vacation in sunny Arizona flying drones (here), I am back soliciting ideas from our Merck Genetic & Pharmacogenomics (GpGx) team. This week’s pick riffs off the events at J.P.Morgan 2015, where there were a number of interesting deals made by pharmaceutical companies and genetic companies (see here, here, here).
With all of this interest in human genetics, it raises the question about how genetics can be used to develop new drugs. The first step is to go from “genes to screens”. That is, the first step is to progress from a human genetic variant associated with a clinical trait of interest to an actual drug screen. This week’s article, published in Nature Chemical Biology, describes one example (see here, here).
Summary of the manuscript: Deleterious mutations in the ABHD12 gene cause a rare neuroinflammatory-neurodegenerative disorder named polyneuropathy, hearing loss, ataxia, retinitis pigmentosa and cataract (PHARC, see here). A similar phenotype is observed in ABHD12-deficient mice. ABHD12 is an enzyme degrading lysophosphatidylserine (lyso-PS), a signaling lipid known to regulate macrophage activation. The Nature Chemical Biology study by Kamat and colleagues describes the chemical proteomic identification of a related enzyme, ABHD16A, which synthesizes the terminal step leading to lyso-PS generation.…
I got a drone for Christmas. The first thing my wife asked me was, “Why do you need a drone?” I did not have a great answer, other than to say it would be fun to take aerial videos. My sister teased me, as did my kids, nieces and nephew (Sam Sutherland). They said I was obsessed; they said I was acting like a little kid. My neighbors were worried – “no more nude sunbathing” was awkwardly expressed by more than one.
The new recreational drones represent pretty cool technology. Just a few years ago, the technology was not available to stabilize and control the flying of drones…at least not at an affordable cost. Now, GPS satellites and gyro sensors can do just that. Until recently, the range on remote controlled drones was relatively limited. Now, wireless communication allows for first-person viewing and long-distance control at long-range (up to 500 meters from my Phantom DJI FC40 drone). And until recently, the cameras attached to drones were not of sufficient quality to record high-definition images. Now, simple microchips installed in HD cameras with stabilizing functions allow for professional-grade photography (e.g., GoPro).
And with these drones, there is a new perspective on old things.…
This week’s theme is genes to function for drug screens…with a macabre theme of zombies! As more genes are discovered through GWAS and large-scale sequencing in humans, there is a pressing need to understand function. There are at least two steps: (1) fine-mapping the most likely causal genes and causal variants; and (2) functional interrogation of causal genes and causal variants to move towards a better understanding of causal human biology for drug screens (“from genes to screens”).
Genome-editing represents one very powerful tool, and the latest article from the laboratory of Feng Zhang at the Broad Institute takes genome-editing to a new level (see Genetic Engineering & Biotechnology News commentary here). They engineer the dead!
Since its introduction in late 2012, the CRISPR-Cas9 gene-editing technology has revolutionized the ways scientists can apply to interrogate gene functions. Using a catalytically inactive Cas9 protein (dead Cas9, dCas9) tethered to an engineered single-guide RNA (sgRNA) molecule, the authors demonstrated the ability to conduct robust gain-of-function genetic screens through programmable, targeted gene activation.
Earlier this year, the laboratories of Stanley Qi, Jonathan Weissman and others \ reported the use of dCas9 conjugated with a transcriptional activator for gene activation (see Cell paper here).…
Welcome to this second blog post on genetics/genomics for drug discovery! So far, we are 2 for 2. That is, this is the second week in a row where we have reviewed the literature for interesting journal articles and written a blog on why the study is relevant for drug discovery. I say “we”, because this week I asked for input from our Merck Genetic & Pharmacogenomics (GpGx) team. We received a number of interesting submissions from GpGx team members, as summarized at the end of the blog.
This week’s article uses antisense as therapeutic proof-of-concept in humans for a genetic target…again! This story is reminiscent of last week’s post on APOC3 (see here).
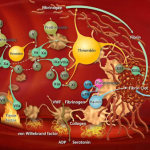
Summary of the manuscript: While patients with congenital Factor XI deficiency have a reduced risk of venous thromboembolism (VTE), it is unknown whether therapeutic modulation of Factor XI will prevent venous thromboembolism without increasing the risk of bleeding. In this open-label, parallel-group study, 300 patients who were undergoing elective primary unilateral total knee arthroplasty were randomly assigned to receive one of two doses of FXI-ASO (200 mg or 300 mg) or 40 mg of enoxaparin once daily.…
There are an increasing number of very interesting published studies around genetics / genomics and drug discovery. Just last week, there were a series of articles in Nature on predictors of response to anti-PD1 therapy. (Disclaimer: I work for Merck, which markets an anti-PD1 drug.) In this new blog series, I try and pick at least one paper that highlights some of the key principles of genetics/genomics and drug discovery. This is week #1…hopefully I will be able to do this routinely!
This week, I have selected an article in NEJM by Gaudet et al: Targeting APOC3 in the Familial Chylomicronemia Syndrome. As described in the introduction: “The familial chylomicronemia syndrome is a rare autosomal recessive disease characterized by the buildup in the blood of fat particles called chylomicrons (chylomicronemia), severe hypertriglyceridemia, and the caused by mutations in the gene encoding LPL or, less frequently, by mutations in genes encoding other proteins necessary for LPL function. Patients with this syndrome have plasma triglyceride levels ranging from 10 to 100 times the normal value (1500 to 15,000 mg per deciliter [17 to 170 mmol per liter]), eruptive xanthomas, arthralgias, neurologic symptoms, lipemia retinalis, and hepatosplenomega without pancreatitis, that interfere with normal life and result in frequent hospitalizations.”…
At the Harvard-Partners Personalized Medicine Conference last week I participated in a panel discussion on complex traits. When asked about where personalized medicine for complex traits will be in the future, I answered that I envision two major categories for personalized therapies.
(1)Development of drugs based on genetic targets will lead to personalized medicine; and
(2)Large effect size variants will be detected in clinical trials or in post-approval studies and will lead to personalized medicine.
This answer, I said, was based in part on current categories of FDA pharmacogenetic labels and in part on how I see new drug discovery occurring in the future. But did the current FDA labels really support this view?
The answer is “yes”. In reviewing the 158 FDA labels (Excel spreadsheet here), my crude analysis found that 31% of labels fall into the “genetic target” category (most from oncology – 26% of total) and 65% fall into the “large effect” category (most from drug metabolism [42% of total], HLA or G6PD [15% of total]).
A subtle but important point is that I predict that category #2 (PGx markers for non-oncology “genetic targets”) will grow in the future. In other words, development of non-oncology drugs will riff-off the success of drugs developed based on somatic cell genetics in oncology. …
I have come across three reports in the last few days that help me think about the question: How many genomes is enough? My conclusion – we need a lot! Here are some thoughts and objective data that support this conclusion.
(1) Clinical sequencing for rare disease – JAMA reported compelling evidence that exome sequencing identified a molecular diagnosis for patients (Editorial here). One study investigated 2000 consecutive patients who had exome sequencing at one academic medical center over 2 years (here). Another study investigated 814 consecutive pediatric patients over 2.5 years (here). Both groups report that ~25% of patients were “solved” by exome sequencing. All patients had a rare clinical presentation that strongly suggested a genetic etiology.
(2) Inactivating NPC1L1 mutations protect from coronary heart diease – NEJM reported an exome sequencing study in ~22,000 case-control samples to search for coronary heart disease (CHD) genes, with follow-up of a specific inactivating mutation (p.Arg406X in the gene NPC1L1) in ~91,000 case-control samples (here). The data suggest that naturally occurring mutations that disrupt NPC1L1 function are associated with reduced LDL cholesterol levels and reduced risk of CHD. The statistics were not overwhelming despite the large sample size (P=0.008, OR=0.47). …
So, you have a target and want to start a drug discovery program, do ya? How would you do it?
When I was at Brigham and Women’s Hospital, Harvard Medical School and the Broad Institute, I presented an idea from an early GWAS of rheumatoid arthritis (RA, see here) to Ed Scolnick (former president of Merck Research Labs, now founding director of the Stanely Center at the Broad Institute, see here). In this study, we found evidence that a non-coding variant at the CD40 gene locus increased risk of RA. The first questions he asked: How does the genetic mutation alter CD40 function? Is it gain-of-function or loss-of-function? What assay would you use for a high-throughput small molecule screen to recapitulate the genetic finding?
I was caught off-guard. Sadly, I had never really thought about all of the details. At the time, I knew enough as a clinician, biologist and a geneticist to appreciate that CD40 was an attractive drug target for RA. However, I was quite naïve to the steps required to take a target into a drug screen. That simple conversation led to several years worth of work, which ultimately led to a proof-of-concept phenotypic screen published in PLoS Genetics five years later (see here).…
In my previous blog series I talked about why genetics is important in drug discovery: human genetics takes you to a target, informs on mechanism of action (MOA) for therapeutic perturbation, provides guidance for pre-clinical assays of target engagement, and facilitates indication selection for clinical trials.
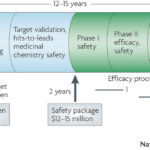
Here, I provide an overview of a new blog series on how genetics influences decision-making during drug discovery. The key principle: human genetics establishes a disciplined mindset and a firm foundation – anchoring points – for advancing targets through the complicated process of drug discovery. [For those less familiar with drug discovery, the end of this blog provides a brief primer on the stages of drug discovery.]
I highlight three areas: establishing a balanced portfolio, identifying targets with novel MOA, and creating a framework for objective decision-making. In subsequent posts, I will focus primarily on how human genetics informs on the latter (decision-making), with blogs pertaining to designing assays for screens and target engagement, utilizing pre-clinical animal models, predicting on-target adverse drug events, and selecting indications for clinical trials.
1. Establish a balanced portfolio
Whether in academic research, a small biotech company (see here) or a large pharmaceutical company (such as Merck, where I work), a balanced portfolio of projects is very important.…
In this post I will build on previous blogs (here, here, here) about genetics for target ID and validation (TIDVAL). Here, I argue that new targets with unambiguous promotable advantage will emerge from studies that focus on genetic pathways rather than single genes.
This is not meant to contradict my previous post about the importance of genetic studies of single genes to identify new targets. However, there are important assumptions about the single gene “allelic series” approach that remain unknown, which ultimately may limit its application. In particular, how many genes exist in the human genome have a series of disease-associated alleles? There are enough examples today to keep biopharma busy. Moreover, I am quite confident that with deep sequencing in extremely large sample sizes (>100,000 patients) such genes will be discovered (see PNAS article by Eric Lander here). Given the explosion of efforts such as Genomics England, Sequencing Initiative Suomi (SISu) in Finland, Geisinger Health Systems, and Accelerating Medicines Partnership, I am sure that more detailed genotype-phenotype maps will be generated in the near future.
[Note: Sisu is a Finnish word meaning determination, bravery, and resilience; it is about taking action against the odds and displaying courage and resoluteness in the face of adversity. …
A key learning from my time in academia was the value of collaborations. Much of my most enjoyable and productive research was conducted in collaboration with fellow scientists across the globe.
I am pleased to report that industry is no different. After one year working for Merck, I have found that in addition to collaborations across the company ties with external scientific experts focused on advancing programs of interest are actively encouraged.
It is heartening to see how some recent progress in several notable drug development programs is leading to increased excitement around the application of human genetics in identifying human drug targets. As I have previously noted, human genetics can also provide insights to identifying pathways enriched for approved drugs (see Nature article here), which indicates that novel pathways may provide an important foundation for novel drug discovery programs. Indeed, the use of pathway-based approaches, including phenotypic screens, can provide a powerful way to make complex genetic pathways actionable for drug discovery.
Today, I am excited to note that Merck has launched a Merck Innovation Network (MINt) Request for Proposals to identify collaborations with academic scientists to evaluate genetic targets or genetic pathways for their potential to become drug discovery programs. …
Question: What can we learn from Sputnik (see here), DARPA (see here) and disruptive innovation (see here) to invent new drugs?
Answer: The best way to prevent surprise is to create it. And if you don’t create the surprise, someone else will. (This is a cryptic answer, I know, but I hope the answer will become clearer by the end of the blog.)
My previous blogs highlighted (1) the pressing need to match an innovative R&D culture with an innovative R&D strategy rooted in basic science (see here), and (2) the importance of phenotype in target ID and validation (TIDVAL) efforts anchored in human genetics (see here). Now, I want to flesh out more of the scientific strategy around human genetics – with a focus on single genes and single drug targets.
To start, I want to frame the problem using an unexpected source of innovation: the US government.
There is an interesting article in Harvard Business Review on DARPA and “Pasteur’s Quadrant” – use-inspired, basic-science research (see here and here). This theme is critically important for drug discovery, as the biopharma industry has a profound responsibility to identify new targets with increased probability-of-success and unambiguous promotable advantage (see here). …
1.The observable physical or biochemical characteristics of an organism, as determined by both genetic makeup and environmental influences.
2. An individual or group of organisms exhibiting a particular phenotype.
There are many different phenotypes: strength in the face of adversity (see here); self-reflection in a time of uncertainty (see here); and creativity amidst a sea of sameness (see here).
Phenotypes also refer to disease states such as risk of disease, response to therapy, a quantitative biomarker of medical relevance, or a physical trait such as height (as in the figure above).
For drug discovery, I have put forth the premise that human genetics is a useful tool to uncover novel drug targets that are likely to have unambiguous promotable advantage (see here). The starting point in a genetic study is to pick the right phenotype, one that is an appropriate surrogate for drug efficacy.
And phenotype matters!
Two illustrative examples are the autoimmune diseases type 1 diabetes and rheumatoid arthritis. In type 1 diabetes the immune system destroys the pancreas, thereby preventing insulin secretion and the control of blood glucose levels.
Human genetics has identified many alleles associated with the risk of type 1 diabetes, nearly all of which act on the immune system (see here). …
The key is to find targets with novel mechanism of action (MOA) and an increased probability of success to differentiate in the clinic.
The pharmaceutical industry is in desperate need of new therapies with “unambiguous promotable advantage” that address unmet clinical need (see here, here and here). Of course, this is a laudable goal in drug development. In fact, given the current health care climate, we have no other choice (see here). If we are to have a sustainable industry, we must change the way we do discovery science. According to the Bernstein Report on BioBusiness: “Differentiation or Bust: Drug companies must start creating the case for value differentiation in discovery and then steadily build a body of evidence throughout the product development process.”
This means that dedicated drug hunters have a steep challenge ahead: to identify targets with novel mechanisms of action that have an increased probability to differentiate in the clinic. This will take creativity, hard work and innovation.
There is a lot written about “innovation”. [See here for a collection of articles from the HBR Insight Center; you can test your “innovation quotient” here.] Most comments about innovation involve creating a climate of risk taking balanced with accountability. …
I believe that humans represent the ideal model organism for the development of innovative therapies to improve human health. Experiments of nature (e.g., human genetics) and longitudinal observations in patients with disease can differentiate between cause and consequence, and therefore can overcome fundamental challenges of drug development (e.g., target identification, biomarkers of drug efficacy). Using my Twitter account (@rplenge), this blog (www.plengegen.com/blog), and other forms of social media, I provide compelling examples that illustrate key concepts of “humans as the ideal model organism” (#himo) for drug development.
Why do drugs fail (#whydrugsfail)? This simple question is at the center of problems facing the pharmaceutical industry. In short, drugs fail in early development because of unresolved safety signals or lack of biomarkers for target engagement, and drugs fail in late development because of lack of efficacy or excess toxicity. This leads to a costly system for bringing new drugs to market – not because of the successes, but because >95% of drug programs ultimately fail. Without improvements in rates of success in drug development, the sustainability of the pharmaceutical industry as we know it is in trouble (see here). Not surprisingly, much has been written about this topic, including analyses of development strategies (Forbes blog, Drug Baron), company pipelines (Nature Reviews Drug Discovery manuscript from AstraZeneca) and FDA approvals (here and here).…
In science, a pendulum swings as new discoveries are made and old hypotheses proven false. Unfortunately, the arc of this swing is often unrelated to the facts, but more tied to the prevailing views of what is and what should be. With incomplete information, the pendulum may swing too far in one direction – for example, towards the view that genome-wide association studies (GWAS) will identify the vast majority of genetic risk for complex traits in relatively small cohorts (now defined humbly as tens-of-thousands of case-control samples). After an initial wave of discoveries – or lack thereof – the pendulum swung too far in the other direction: disease-associated variants from GWAS cannot explain most of the estimated heritability in complex traits, therefore rare variants of large effect must be the root genetic cause of complex traits.
Too often, science creates an artificial mirror image of data interpretation. If one hypothesis is not true, then the opposite must be true. If it is not common variants, then it must be rare variants; if it is not genetics, then it must be epigenetics; if it is not the host, then it must be the microbiome; and so forth. Too often, incomplete data to support one model results in a knee-jerk reaction towards an orthogonal model, even if there is little evidence to support the model. …
As I sought advice from colleagues about my career, I was frequently asked if I would prefer to work in academics or industry (emphasis on the word “or”). The standard discussion went something like this:
ACADEMICS – you are your own boss and you are free to chose your own scientific direction; funding is tight, but good science still gets funded by the NIH, foundations and other organizations (including industry); the team unit centers around individuals (graduate students, post-docs, etc), which favors innovative science but sometimes makes large, multi-disciplinary projects challenging; there is long-term stability, including control over where you want to work and live, assuming funding is procured and good ideas continue; your base salary will be less than in industry, but you still make a good living and there are opportunities to consult – and maybe even start your own company – to supplement income. Bottom line: if you want to do innovative science under your own control, work in academics – as that is where most fundamental discoveries are made.
INDUSTRY – there are more resources, but those resources are not necessarily under your control (depending upon your seniority); the company may change direction quickly, which changes what you are able to work on; while drug development takes 10-plus years, many goals are short-term (several years), which limits long-term investment in projects that are risky and require years to develop; the team unit centers around projects (e.g.,…
Bill James developed the “Keltner list” to serve as a series of gut-check questions to test a baseball player’s suitability for the Hall of Fame (see here). The list comprises 15 questions designed to aid in the thought process, where each question is designed to be relatively easy to answer. As a subjective method, the Keltner list is not designed to yield an undeniable answer about a player’s worthiness. Says James: “You can’t total up the score and say that everybody who is at eight or above should be in, or anything like that.”
The Keltner list concept has been adapted to address to serve as a common sense assessment of non-baseball events, including political scandals (see here) and rock bands like Devo (see here).
Here, I try out this concept for genetics and drug discovery. That is, I ask a series of question designed to answer the question: “Would a drug against the product of this gene be a useful drug?” I use PCSK9 as one of the best examples (see brief PCSK9 slide deck here). I also used in on our recent study of CD40 in rheumatoid arthritis, published in PLoS Genetics (see here).…
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}