

A new sickle cell anemia gene therapy study published in the New England Journal of Medicine (see here, here) gives hope to patients and the concept of rapidly programmable therapeutics based on causal human biology. But how close are we really?
It takes approximately 5-7 years to advance from a therapeutic hypothesis to an early stage clinical trial, and an additional 4-7 years of late stage clinical studies to advance to regulatory approval. This is simply too long, too inefficient and too expensive.
But how can timelines be shortened?
In the current regulatory environment, it is difficult to compress late stage development timelines. This leaves the time between target selection (or “discovery”) and early clinical trials (ideally clinical proof-of-concept, or “PoC”) as an important time to gain efficiencies. Further, discovery to PoC is an important juncture for minimizing failure rates in late development and delivering value to patients in the real world (see here).
Here, I argue that rapidly programmable therapeutics based a molecular understanding of the causal disease process is key to compressing the discovery to PoC timeline.
Imagine a world where the molecular basis of disease is completely understood. For common diseases, germline genetics contributes approximately two-thirds of risk; for rare diseases, germline genetics contributes nearly 100% of risk. For cancer, somatic genetic alterations and the interplay with the immune system are major drivers of disease. For infectious diseases, host-pathogen interactions lead to an antigen-directed immune response that clears the invading microbe.
Thanks to advances in genetics, genomics and the human immune system, science is making progress towards understanding the molecular basis of common/rare diseases, cancer and infections (e.g., see this week’s GRAIL announcement here, where circulating tumor DNA can be used to detect cancer at an early stage). In contrast, progress has been slow at creating therapeutic molecules that reverse disease processes and that can and then be tested in humans for PoC.
And here is where the NEJM study by Ribeil et al is important.
The first insight into the molecular basis of sickle cell anemia came back in 1956 (here). Yep, that is more than 60 years ago, the same year that Elvis Presley first appeared on The Ed Sullivan Show and IBM produced a 5 MB computer hard drive. This timeline is a reminder that understanding the genetic basis of disease is only one small step toward intercepting, preventing or curing disease.
Rebeil et al, working with a team from bluebird bio ($BLUE), devised a gene therapy vector system to permanently introduce a normal copy of the mutated gene, beta-globin, into the bone marrow of a patient with sickle cell anemia. Their gene therapy and stem cell transplant approach works as follows: (a) bone marrow biopsy to harvest stem cells; (b) ex vivo transfection of CD34+ stem cells with antisickling β-globin variant in a lentiviral gene therapy backbone; (c) myeloablation with intravenous busulfan to remove endogenous stem cells; and (d) autologous transplant of genetically modified stem cells. (See useful YouTube videos here and here, which are also on the bluebird bio website.)
And it worked.
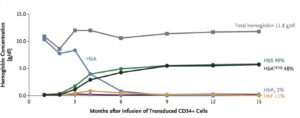
The transplanted, genetically modified CD34+ stem cells began to produce normal red blood cells after about 3 months (see Figure 1, below). The patient was discharged from the hospital on day 50. The last transfusion was 88 days after transplant. More than 15 months after transplantation, no sickle cell disease–related clinical events or hospitalization had occurred.
How might this advance lead to programmable therapeutics?
When writing code (i.e., computer programming), it is possible to iterate quickly. In a short period of time, it is possible to define a problem, write code to solve the problem, and iterate in real-time as issues arise.
Imagine if that same system were in place for therapeutics: a molecular defect is diagnosed, a programmable therapeutic is taken off the shelf and introduced safely into humans, and the therapeutic is tweaked (e.g., dose, duration) to achieved the desired therapeutic margin based on clinical data in humans.
We are a long way off from this reality, but advances such as that described by Rebeil et al make it seem possible. In addition to gene therapy with endogenous lentiviral vectors, substantial progress is being made in CRISPR/cas9 gene editing (see here), mRNA (see here), oligonucleotide, biologic, cellular, and microbiome therapeutics. In theory, it should be possible to devise a system to rapidly program a therapeutic modality to reverse an underlying molecular defect.
I conclude by saying that programmable therapeutics based on causal human biology is not simply an aspiration…it is a necessity. The drug R&D process is too slow, too expensive and too inefficient to be sustainable. Something needs to change, and the concept of programmable therapeutics – as far off as it may seem today – is essential.
I would also argue that it is feasible to compress this timeline to less than one year. Sure, that may seem impossible today. But back in 1956 when Elvis was groovin’ on TV and computer geeks were salivating over 5 MB hard drives, did anyone think we would have live streaming of anything by anybody across the entire world? Why not molecular defects to human PoC via programmable therapeutics in less than a year!