At the Harvard-Partners Personalized Medicine Conference last week I participated in a panel discussion on complex traits. When asked about where personalized medicine for complex traits will be in the future, I answered that I envision two major categories for personalized therapies.
(1)Development of drugs based on genetic targets will lead to personalized medicine; and
(2)Large effect size variants will be detected in clinical trials or in post-approval studies and will lead to personalized medicine.
This answer, I said, was based in part on current categories of FDA pharmacogenetic labels and in part on how I see new drug discovery occurring in the future. But did the current FDA labels really support this view?
The answer is “yes”. In reviewing the 158 FDA labels (Excel spreadsheet here), my crude analysis found that 31% of labels fall into the “genetic target” category (most from oncology – 26% of total) and 65% fall into the “large effect” category (most from drug metabolism [42% of total], HLA or G6PD [15% of total]).
A subtle but important point is that I predict that category #2 (PGx markers for non-oncology “genetic targets”) will grow in the future. In other words, development of non-oncology drugs will riff-off the success of drugs developed based on somatic cell genetics in oncology. …
I have come across three reports in the last few days that help me think about the question: How many genomes is enough? My conclusion – we need a lot! Here are some thoughts and objective data that support this conclusion.
(1) Clinical sequencing for rare disease – JAMA reported compelling evidence that exome sequencing identified a molecular diagnosis for patients (Editorial here). One study investigated 2000 consecutive patients who had exome sequencing at one academic medical center over 2 years (here). Another study investigated 814 consecutive pediatric patients over 2.5 years (here). Both groups report that ~25% of patients were “solved” by exome sequencing. All patients had a rare clinical presentation that strongly suggested a genetic etiology.
(2) Inactivating NPC1L1 mutations protect from coronary heart diease – NEJM reported an exome sequencing study in ~22,000 case-control samples to search for coronary heart disease (CHD) genes, with follow-up of a specific inactivating mutation (p.Arg406X in the gene NPC1L1) in ~91,000 case-control samples (here). The data suggest that naturally occurring mutations that disrupt NPC1L1 function are associated with reduced LDL cholesterol levels and reduced risk of CHD. The statistics were not overwhelming despite the large sample size (P=0.008, OR=0.47). …
I believe that humans represent the ideal model organism for the development of innovative therapies to improve human health. Experiments of nature (e.g., human genetics) and longitudinal observations in patients with disease can differentiate between cause and consequence, and therefore can overcome fundamental challenges of drug development (e.g., target identification, biomarkers of drug efficacy). Using my Twitter account (@rplenge), this blog (www.plengegen.com/blog), and other forms of social media, I provide compelling examples that illustrate key concepts of “humans as the ideal model organism” (#himo) for drug development.
Why do drugs fail (#whydrugsfail)? This simple question is at the center of problems facing the pharmaceutical industry. In short, drugs fail in early development because of unresolved safety signals or lack of biomarkers for target engagement, and drugs fail in late development because of lack of efficacy or excess toxicity. This leads to a costly system for bringing new drugs to market – not because of the successes, but because >95% of drug programs ultimately fail. Without improvements in rates of success in drug development, the sustainability of the pharmaceutical industry as we know it is in trouble (see here). Not surprisingly, much has been written about this topic, including analyses of development strategies (Forbes blog, Drug Baron), company pipelines (Nature Reviews Drug Discovery manuscript from AstraZeneca) and FDA approvals (here and here).…
As I sought advice from colleagues about my career, I was frequently asked if I would prefer to work in academics or industry (emphasis on the word “or”). The standard discussion went something like this:
ACADEMICS – you are your own boss and you are free to chose your own scientific direction; funding is tight, but good science still gets funded by the NIH, foundations and other organizations (including industry); the team unit centers around individuals (graduate students, post-docs, etc), which favors innovative science but sometimes makes large, multi-disciplinary projects challenging; there is long-term stability, including control over where you want to work and live, assuming funding is procured and good ideas continue; your base salary will be less than in industry, but you still make a good living and there are opportunities to consult – and maybe even start your own company – to supplement income. Bottom line: if you want to do innovative science under your own control, work in academics – as that is where most fundamental discoveries are made.
INDUSTRY – there are more resources, but those resources are not necessarily under your control (depending upon your seniority); the company may change direction quickly, which changes what you are able to work on; while drug development takes 10-plus years, many goals are short-term (several years), which limits long-term investment in projects that are risky and require years to develop; the team unit centers around projects (e.g.,…
At the Spring PGRN meeting last week, there were a number of interesting talks about the need for new databases to foster genetics research. One talk was from Scott Weiss on Gene Insight (see here). I gave a talk about our “RA Responder” Crowdsourcing Challenge (complete slide deck here). Here are a few general thoughts about the databases we need for genomics research.
(1) Silo’s are so last year
Too often, data from one interesting pharmacogenomic study (e.g., GWAS data on treatment response) are completely separate from another dataset that can be used to interpret the data (e.g., RNA-sequencing). Yes, specialized labs that generated the data can integrate the data for their own analysis. And yes, they can release individual datasets into the public for others to stitch together. But is this really what we need? Somehow, we need to make data available in a manner that is fully integrated and interoperable. One simple example of this is GWAS for autoimmune diseases. Since 2006, a large number of genetic data have been published. Still, there is no single place to go see results for all autoimmune diseases, despite the fact that there is tremendous shared overlap among the genetic basis for these diseases.…
I read with interest a recent publication by Khandpur et al in Science Translational Medicine on NETosis in the pathogenesis of rheumatoid arthritis (download PDF here). It made me think about “cause vs consequence” in scientific discovery. That is, how does one determine whether a biological process observed in patients with active disease is a cause of disease rather than a consequence of disease?
In reading the article, I learned about how neutrophils cause tissue damage and promote autoimmunity through the aberrant formation of neutrophil extracellular traps (NETs). Released via a novel form of cell death called NETosis, NETs consist of a chromatin meshwork decorated with antimicrobial peptides typically present in neutrophil granules. (Read more about NETs on Wikipedia here.)
Mendelian randomization is a method of using measured variation in genes of known function to examine the causal effect of a modifiable exposure on disease in non-experimental studies (read more here). It is a powerful to determine if an observation in patients is causal. For example, if autoantibodies are pathogenic in RA, then DNA variants that influence the formation of autoantibodies should also be associated with risk of RA. This is indeed the case, as exemplified by variants in a gene, PADI4, the codes for an enzyme involved in peptide citrullination (see here). …
For our website, we have chosen the term “precision medicine” rather than “personalized medicine”. A recent News article in Nature Medicine reinforces this concept (see here).
I have had many of my non-genetic physician colleagues comment to me: “We practice personalized medicine every day. It’s called basic patient care!” Their point: physicians see patients and make decisions about the best course of treatment based on patient preferences. For example, one RA patient may prefer to have a drug infusion once per month and another patient may prefer to take a pill each day.
The Nature Medicine article emphasizes “the idea that molecular information improves the precision with which patients are categorized and treated“. While personalized medicine might say “patient X with disease Y should get drug Z”, precision medicine says “patient X has a subset of disease Y — actually, disease Y3, not disease Y1, Y2 or Y4 — and patients with disease Y tend to respond more favorably to drug Z”. Said another way bt Charles Sawyers, an oncologist at the Memorial Sloan-Kettering Cancer Center in New York: “we are trying to convey a more precise classification of disease into subgroups that in the past have been lumped together because there wasn’t a clear way to discriminate between them“.…
The value of genetics to clinical prediction depends upon the underlying genetic architecture of complex traits (including disease risk and drug efficacy/toxicity). It is increasingly clear that common variants contribute to common phenotypes, but that extremely large sample sizes are required to tease apart true signal from the noise at a stringent level of statistical significance. Occasionally, common variants have a large effect on common phenotypes (e.g., MHC alleles and risk of autoimmunity; VKORC1 and warfarin metabolism), but this seems to be the exception rather than the rule.
A recent paper published in Nature Genetics explores this concept in more detail (download PDF here). As stated in the manuscript by Chatterjee and colleagues: “The gap between estimates of heritability based on known loci and those estimated owing to the comprehensive set of common susceptibility variants raises the possibility of substantially improving prediction performance of risk models by using a polygenic approach, one that includes many SNPs that do not reach the stringent threshold for genome-wide significance.” They measure the ability of models based on current as well as future GWAS to improve the prediction of individual traits.
The results, which are intriguing, depend not only on the underlying genetic architecture (which is often unknown, especially for PGx traits), but also disease prevalence and familial aggregation: “We observed that for less common, highly familial conditions, such as T1D and Crohn’s disease, risk models that include family history and optimal polygenic scores based on current GWAS can identify a large majority of cases by targeting a small group of high-risk individuals (for example, subjects who fall in the highest quintile of risk).…
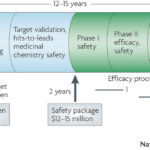
Genetics can guide the first phase of drug development (identifying drug targets, see here ) as well as late phase clinical trials (e.g., patient segmentation for response/non-responder status, see here ). But is there a convergence between the two areas, or pharmaco-convergence (a term I just made up!)? And are there advantages to a program anchored at both ends in human genetics?
Consider the following two hypothetical examples.
(1) Human genetics identifies loss-of-function (LOF) mutations that protect from disease. The same LOF mutation is associated with an intermediate biomarker, but is not associated with other phenotypes that might be considered adverse drug events. A drug is developed that mimics the effect of the mutation; that is, a drug is developed that inhibits the protein product of the gene. In early mechanistic studies, the drug is shown to influence the intermediate biomarker in a way that is consistent to that predicted by the LOF-protective mutations. Further, because functional studies of the LOF-protective mutations provide insight into relevant biological pathways in humans (e.g., a gene expression signature that correlates with mutation carrier status), additional information is known about genomic signatures of those who carry the LOF-protective mutations (which mimics drug exposure) compared to those who do not carry the LOF-protective mutations (which mimics those who are not exposed to drug).…
Are the same standards applied to genetic and non-genetic tests in clinical medicine? In a review by Munir Pirmohamed and Dyfrig Hughes (download PDFhere), the authors “strongly argue that the slow progress in the implementation of pharmacogenetic (and indeed other genetic) tests can partly be explained by the fact that different criteria are applied when considering genetic testing compared with non-genetic diagnostic tests.” They provide a few compelling examples:
(1) Atomoxetine
There is no regulatory requirement to undertake clinical trials to show that the dosing recommendations for patients with, for example, renal impairment are equivalent in terms of clinical outcomes to those for patients with normal renal function. Indeed, such a stipulation would be impractical and costly, and would never be done during the drug development process, potentially disadvantaging vulnerable patient populations.
Atomoxetine, a drug widely used for attention deficit hyperactivity disorder, is metabolized in the liver by CYP2D6. The SmPC for atomoxetine states that the dose should be reduced by 50% in patients with hepatic impairment (Child-Pugh class B), as drug exposure goes up by twofold. It is also known that drug exposure is increased by a similar amount in CYP2D6 PMs; however, although the SmPC for atomoxetine mentions the effect of CYP2D6 polymorphisms, it does not mandate testing for their presence.…










{kind=link}
{kind=link}
{kind=link}
{kind=link}