1.The observable physical or biochemical characteristics of an organism, as determined by both genetic makeup and environmental influences.
2. An individual or group of organisms exhibiting a particular phenotype.
There are many different phenotypes: strength in the face of adversity (see here); self-reflection in a time of uncertainty (see here); and creativity amidst a sea of sameness (see here).
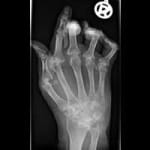
Phenotypes also refer to disease states such as risk of disease, response to therapy, a quantitative biomarker of medical relevance, or a physical trait such as height (as in the figure above).
For drug discovery, I have put forth the premise that human genetics is a useful tool to uncover novel drug targets that are likely to have unambiguous promotable advantage (see here). The starting point in a genetic study is to pick the right phenotype, one that is an appropriate surrogate for drug efficacy.
And phenotype matters!
Two illustrative examples are the autoimmune diseases type 1 diabetes and rheumatoid arthritis. In type 1 diabetes the immune system destroys the pancreas, thereby preventing insulin secretion and the control of blood glucose levels.
Human genetics has identified many alleles associated with the risk of type 1 diabetes, nearly all of which act on the immune system (see here). …
I believe that humans represent the ideal model organism for the development of innovative therapies to improve human health. Experiments of nature (e.g., human genetics) and longitudinal observations in patients with disease can differentiate between cause and consequence, and therefore can overcome fundamental challenges of drug development (e.g., target identification, biomarkers of drug efficacy). Using my Twitter account (@rplenge), this blog (www.plengegen.com/blog), and other forms of social media, I provide compelling examples that illustrate key concepts of “humans as the ideal model organism” (#himo) for drug development.
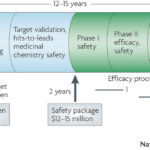
Why do drugs fail (#whydrugsfail)? This simple question is at the center of problems facing the pharmaceutical industry. In short, drugs fail in early development because of unresolved safety signals or lack of biomarkers for target engagement, and drugs fail in late development because of lack of efficacy or excess toxicity. This leads to a costly system for bringing new drugs to market – not because of the successes, but because >95% of drug programs ultimately fail. Without improvements in rates of success in drug development, the sustainability of the pharmaceutical industry as we know it is in trouble (see here). Not surprisingly, much has been written about this topic, including analyses of development strategies (Forbes blog, Drug Baron), company pipelines (Nature Reviews Drug Discovery manuscript from AstraZeneca) and FDA approvals (here and here).…
In science, a pendulum swings as new discoveries are made and old hypotheses proven false. Unfortunately, the arc of this swing is often unrelated to the facts, but more tied to the prevailing views of what is and what should be. With incomplete information, the pendulum may swing too far in one direction – for example, towards the view that genome-wide association studies (GWAS) will identify the vast majority of genetic risk for complex traits in relatively small cohorts (now defined humbly as tens-of-thousands of case-control samples). After an initial wave of discoveries – or lack thereof – the pendulum swung too far in the other direction: disease-associated variants from GWAS cannot explain most of the estimated heritability in complex traits, therefore rare variants of large effect must be the root genetic cause of complex traits.
Too often, science creates an artificial mirror image of data interpretation. If one hypothesis is not true, then the opposite must be true. If it is not common variants, then it must be rare variants; if it is not genetics, then it must be epigenetics; if it is not the host, then it must be the microbiome; and so forth. Too often, incomplete data to support one model results in a knee-jerk reaction towards an orthogonal model, even if there is little evidence to support the model. …
As I sought advice from colleagues about my career, I was frequently asked if I would prefer to work in academics or industry (emphasis on the word “or”). The standard discussion went something like this:
ACADEMICS – you are your own boss and you are free to chose your own scientific direction; funding is tight, but good science still gets funded by the NIH, foundations and other organizations (including industry); the team unit centers around individuals (graduate students, post-docs, etc), which favors innovative science but sometimes makes large, multi-disciplinary projects challenging; there is long-term stability, including control over where you want to work and live, assuming funding is procured and good ideas continue; your base salary will be less than in industry, but you still make a good living and there are opportunities to consult – and maybe even start your own company – to supplement income. Bottom line: if you want to do innovative science under your own control, work in academics – as that is where most fundamental discoveries are made.
INDUSTRY – there are more resources, but those resources are not necessarily under your control (depending upon your seniority); the company may change direction quickly, which changes what you are able to work on; while drug development takes 10-plus years, many goals are short-term (several years), which limits long-term investment in projects that are risky and require years to develop; the team unit centers around projects (e.g.,…
Bill James developed the “Keltner list” to serve as a series of gut-check questions to test a baseball player’s suitability for the Hall of Fame (see here). The list comprises 15 questions designed to aid in the thought process, where each question is designed to be relatively easy to answer. As a subjective method, the Keltner list is not designed to yield an undeniable answer about a player’s worthiness. Says James: “You can’t total up the score and say that everybody who is at eight or above should be in, or anything like that.”
The Keltner list concept has been adapted to address to serve as a common sense assessment of non-baseball events, including political scandals (see here) and rock bands like Devo (see here).
Here, I try out this concept for genetics and drug discovery. That is, I ask a series of question designed to answer the question: “Would a drug against the product of this gene be a useful drug?” I use PCSK9 as one of the best examples (see brief PCSK9 slide deck here). I also used in on our recent study of CD40 in rheumatoid arthritis, published in PLoS Genetics (see here).…
At the Spring PGRN meeting last week, there were a number of interesting talks about the need for new databases to foster genetics research. One talk was from Scott Weiss on Gene Insight (see here). I gave a talk about our “RA Responder” Crowdsourcing Challenge (complete slide deck here). Here are a few general thoughts about the databases we need for genomics research.
(1) Silo’s are so last year
Too often, data from one interesting pharmacogenomic study (e.g., GWAS data on treatment response) are completely separate from another dataset that can be used to interpret the data (e.g., RNA-sequencing). Yes, specialized labs that generated the data can integrate the data for their own analysis. And yes, they can release individual datasets into the public for others to stitch together. But is this really what we need? Somehow, we need to make data available in a manner that is fully integrated and interoperable. One simple example of this is GWAS for autoimmune diseases. Since 2006, a large number of genetic data have been published. Still, there is no single place to go see results for all autoimmune diseases, despite the fact that there is tremendous shared overlap among the genetic basis for these diseases.…
I prepared a lecture for immunology graduate students at Harvard Medical School on clinical features of rheumatoid arthritis (RA) for the G1 IMM302qc class.
This blog post pertains to the Systems Immunology graduate course at Harvard Medical School (Immunology 306qc; see here), which is led by Drs. Christophe Benoist, Nick Haining and Nir Hacohen. My lecture is on the role of human genetics as a tool for understanding the human immune system in health and disease. What follows is an informal description of my lecture. The slide deck for the lecture can be downloaded here. Throughout, I have added key references, with links to the manuscripts and other web-based resources embedded within the blog (and also listed at the end). I highlight five key manuscripts (#1,#2, #3, #4, and #5), which should be reviewed prior to the lecture; the other references, while interesting, are optional.
Overview
It is increasingly clear that humans serve as the best model organism for understanding human health and disease. One reason for this paradigm shift is the lack of fidelity of most animal models to human disease. For systems immunology, the mouse is a powerful model organism to understand fundamental mechanisms of the immune system. However, studies in humans are required to understand how these mechanisms can be translated into new biomarkers and drugs.…
I read with interest a recent publication by Khandpur et al in Science Translational Medicine on NETosis in the pathogenesis of rheumatoid arthritis (download PDF here). It made me think about “cause vs consequence” in scientific discovery. That is, how does one determine whether a biological process observed in patients with active disease is a cause of disease rather than a consequence of disease?
In reading the article, I learned about how neutrophils cause tissue damage and promote autoimmunity through the aberrant formation of neutrophil extracellular traps (NETs). Released via a novel form of cell death called NETosis, NETs consist of a chromatin meshwork decorated with antimicrobial peptides typically present in neutrophil granules. (Read more about NETs on Wikipedia here.)
Mendelian randomization is a method of using measured variation in genes of known function to examine the causal effect of a modifiable exposure on disease in non-experimental studies (read more here). It is a powerful to determine if an observation in patients is causal. For example, if autoantibodies are pathogenic in RA, then DNA variants that influence the formation of autoantibodies should also be associated with risk of RA. This is indeed the case, as exemplified by variants in a gene, PADI4, the codes for an enzyme involved in peptide citrullination (see here). …
The value of genetics to clinical prediction depends upon the underlying genetic architecture of complex traits (including disease risk and drug efficacy/toxicity). It is increasingly clear that common variants contribute to common phenotypes, but that extremely large sample sizes are required to tease apart true signal from the noise at a stringent level of statistical significance. Occasionally, common variants have a large effect on common phenotypes (e.g., MHC alleles and risk of autoimmunity; VKORC1 and warfarin metabolism), but this seems to be the exception rather than the rule.
A recent paper published in Nature Genetics explores this concept in more detail (download PDF here). As stated in the manuscript by Chatterjee and colleagues: “The gap between estimates of heritability based on known loci and those estimated owing to the comprehensive set of common susceptibility variants raises the possibility of substantially improving prediction performance of risk models by using a polygenic approach, one that includes many SNPs that do not reach the stringent threshold for genome-wide significance.” They measure the ability of models based on current as well as future GWAS to improve the prediction of individual traits.
The results, which are intriguing, depend not only on the underlying genetic architecture (which is often unknown, especially for PGx traits), but also disease prevalence and familial aggregation: “We observed that for less common, highly familial conditions, such as T1D and Crohn’s disease, risk models that include family history and optimal polygenic scores based on current GWAS can identify a large majority of cases by targeting a small group of high-risk individuals (for example, subjects who fall in the highest quintile of risk).…








{kind=link}
{kind=link}