I have come across three reports in the last few days that help me think about the question: How many genomes is enough? My conclusion – we need a lot! Here are some thoughts and objective data that support this conclusion.
(1) Clinical sequencing for rare disease – JAMA reported compelling evidence that exome sequencing identified a molecular diagnosis for patients (Editorial here). One study investigated 2000 consecutive patients who had exome sequencing at one academic medical center over 2 years (here). Another study investigated 814 consecutive pediatric patients over 2.5 years (here). Both groups report that ~25% of patients were “solved” by exome sequencing. All patients had a rare clinical presentation that strongly suggested a genetic etiology.
(2) Inactivating NPC1L1 mutations protect from coronary heart diease – NEJM reported an exome sequencing study in ~22,000 case-control samples to search for coronary heart disease (CHD) genes, with follow-up of a specific inactivating mutation (p.Arg406X in the gene NPC1L1) in ~91,000 case-control samples (here). The data suggest that naturally occurring mutations that disrupt NPC1L1 function are associated with reduced LDL cholesterol levels and reduced risk of CHD. The statistics were not overwhelming despite the large sample size (P=0.008, OR=0.47). …
I believe that humans represent the ideal model organism for the development of innovative therapies to improve human health. Experiments of nature (e.g., human genetics) and longitudinal observations in patients with disease can differentiate between cause and consequence, and therefore can overcome fundamental challenges of drug development (e.g., target identification, biomarkers of drug efficacy). Using my Twitter account (@rplenge), this blog (www.plengegen.com/blog), and other forms of social media, I provide compelling examples that illustrate key concepts of “humans as the ideal model organism” (#himo) for drug development.
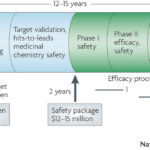
Why do drugs fail (#whydrugsfail)? This simple question is at the center of problems facing the pharmaceutical industry. In short, drugs fail in early development because of unresolved safety signals or lack of biomarkers for target engagement, and drugs fail in late development because of lack of efficacy or excess toxicity. This leads to a costly system for bringing new drugs to market – not because of the successes, but because >95% of drug programs ultimately fail. Without improvements in rates of success in drug development, the sustainability of the pharmaceutical industry as we know it is in trouble (see here). Not surprisingly, much has been written about this topic, including analyses of development strategies (Forbes blog, Drug Baron), company pipelines (Nature Reviews Drug Discovery manuscript from AstraZeneca) and FDA approvals (here and here).…
At the Spring PGRN meeting last week, there were a number of interesting talks about the need for new databases to foster genetics research. One talk was from Scott Weiss on Gene Insight (see here). I gave a talk about our “RA Responder” Crowdsourcing Challenge (complete slide deck here). Here are a few general thoughts about the databases we need for genomics research.
(1) Silo’s are so last year
Too often, data from one interesting pharmacogenomic study (e.g., GWAS data on treatment response) are completely separate from another dataset that can be used to interpret the data (e.g., RNA-sequencing). Yes, specialized labs that generated the data can integrate the data for their own analysis. And yes, they can release individual datasets into the public for others to stitch together. But is this really what we need? Somehow, we need to make data available in a manner that is fully integrated and interoperable. One simple example of this is GWAS for autoimmune diseases. Since 2006, a large number of genetic data have been published. Still, there is no single place to go see results for all autoimmune diseases, despite the fact that there is tremendous shared overlap among the genetic basis for these diseases.…
I read with interest a recent publication by Khandpur et al in Science Translational Medicine on NETosis in the pathogenesis of rheumatoid arthritis (download PDF here). It made me think about “cause vs consequence” in scientific discovery. That is, how does one determine whether a biological process observed in patients with active disease is a cause of disease rather than a consequence of disease?
In reading the article, I learned about how neutrophils cause tissue damage and promote autoimmunity through the aberrant formation of neutrophil extracellular traps (NETs). Released via a novel form of cell death called NETosis, NETs consist of a chromatin meshwork decorated with antimicrobial peptides typically present in neutrophil granules. (Read more about NETs on Wikipedia here.)
Mendelian randomization is a method of using measured variation in genes of known function to examine the causal effect of a modifiable exposure on disease in non-experimental studies (read more here). It is a powerful to determine if an observation in patients is causal. For example, if autoantibodies are pathogenic in RA, then DNA variants that influence the formation of autoantibodies should also be associated with risk of RA. This is indeed the case, as exemplified by variants in a gene, PADI4, the codes for an enzyme involved in peptide citrullination (see here). …




{kind=link}
{kind=link}